13651923355

技术文章
液相色谱 - 质谱(LC-MS)已成为许多具有挑战性的分析的首xuan分析技术,基于其选择性,灵敏度和对不同极性化合物的广泛适用性。尽管该技术具有优势,但LC-MS系统的复杂性常常使分析人员难以满足方法检测限制。在“Column Watch”的这一部分中,讨论了几种策略,通过减少污染物,仔细选择LC方法条件以及优化MS接口设置来提高方法灵敏度。通过了解这些参数与电离效率之间的关系,分析人员可以提高其信噪比并实现LC-MS技术的隐藏潜力。
在质谱(MS)中,术语灵敏度可具有通常可互换使用的若干含义。灵敏度可定义为每单位分析物浓度变化的信号变化(例如校准曲线的斜率)[1]。更常见的是,它用于参考MS检测器中分析物产生的信号的大小。在后一种用法中,MS灵敏度通常用于比较检测器。
从根本上说,检测器提供定量数据的能力是分析物的信噪比(S / N)的函数。检测限(LOD)由分析物S / N确定,是物质的低浓度,其信号可与系统噪声区分开来[2]。如图1所示,如果背景噪声保持不变,则MS的灵敏度越高,给定方法LOD的S / N值越大。因此,通过操纵S / N可以发生灵敏度的提高。MS优化,样品预处理策略,流动相组成和LC色谱柱特性都是电离效率*的,并且在优化时会改善分析物信号。同样,限制有助于信号抑制或加合物形成的污染物也可以增强响应。
图1 假设线性校准和固定背景噪声,假设增加灵敏度对检测限(LOD)的影响。 |
MS优化
在液相色谱 - 质谱(LC-MS)中,灵敏度直接关系到从溶液中的分析物产生气相离子的效率(电离效率)以及将它们从大气压转移到MS系统的低压区的能力。 (传输效率)[3]。电离和传输效率的优化取决于LC方法参数和目标分析物或分析物。为了进行适当的调整,有必要对MS源中发生的机制有基本的了解。
电喷雾电离(ESI)是zui流行的电离技术之一; 因此,它将成为本专栏文章的重点。然而,重要的是要注意,无论选择何种电离模式,都必须优化源参数。当LC流动相流入样品毛细管时,基于所选择的极性分离正离子和负离子。在正ESI中,负离子在毛细管壁上被中和,并且正离子在流动相中继续到毛细管,其中带电分析物累积成液滴。在施加电压的影响下,形成泰勒锥[4]。静电排斥导致锥体破碎成小的带电液滴,然后,在毛细管和采样板之间施加的电位差的引导下,它朝向采样孔行进。随着微小液滴向孔口前进,溶剂在干燥气体和热量的作用下蒸发,导致液滴表面积减小,电荷密度增加。终,排斥力克服液滴表面张力,液滴爆炸成更小的液滴。该过程重复进行,直到液滴太小以至于发射出气相离子[5]。形成的离子云被称为 终,排斥力克服液滴表面张力,液滴爆炸成更小的液滴。该过程重复进行,直到液滴太小以至于发射出气相离子[5]。形成的离子云被称为 终,排斥力克服液滴表面张力,液滴爆炸成更小的液滴。该过程重复进行,直到液滴太小以至于发射出气相离子[5]。形成的离子云被称为离子羽。
选择合适的极性是开发灵敏的LC-MS方法的步。选择毛细管极性以匹配目标分析物的电荷。通常,碱性分析物通过接受质子(M + H)+以正离子模式zui有效地电离,而酸性分析物通过提供质子(MH)-将在负离子模式中产生zui强的信号。然而,对于更复杂的分子,可能难以预测*极性模式。此外,分析物的行为和响应因仪器平台而异。因此,在初始方法开发期间或将现有方法转移到新仪器时,使用两种极性模式筛选分析物是有益的[6]。
电离效率受流速,流动相组成和目标分析物的物理化学性质的强烈影响。毛细管电压设置取决于分析物,洗脱液和流速,并且可能对方法重现性产生重大影响。毛细管和采样板之间施加的电位差是维持稳定和可重复喷雾的原因[7]。如果毛细管电压设置不正确,可能会出现可变电离和精度问题。*雾化气体流量和温度也取决于洗脱液。雾化气体限制了液滴的生长,同时电荷累积并且还影响从毛细管发射的液滴的尺寸。应增加雾化气体流量和温度以获得更快的LC流速或使用高含水流动相时。类似地,干燥气体流量和温度对于LC洗脱液的有效去溶剂化和气相离子的成功生产是关键的。需要注意的是,在分析热不稳定分析物时,必须注意防止其在源中降解。
在电离源内产生气相离子的位置对于*传输到MS系统是重要的。离子羽流的大小取决于发射气相离子所需的裂变事件的数量及其与采样孔的距离。通过基于LC流速调节毛细管相对于孔口的位置,可以优化离子羽流的取样。在更快的流速下,毛细管应放置在离采样孔更远的位置,以便进行充分的去溶剂化和增加裂变事件的数量。尽管延长距离将允许产生更多数量的气相离子,但排斥力也将成比例地增加,导致离子羽流的尺寸扩大并且气相离子的密度减小。结果是,进入采样孔的离子数量会减少,导致信号强度下降[3]。在较慢的流速下,形成较小的液滴,使毛细管更靠近取样孔。较小的液滴更容易去溶剂化并且需要更少的裂变事件,减少排斥力的影响并抑制离子羽流的大小。毛细管和采样孔之间的距离减小会增加离子羽流密度,提高分析物的电离效率和传输效率[3]。减少排斥力的影响并抑制离子羽流的大小。毛细管和采样孔之间的距离减小会增加离子羽流密度,提高分析物的电离效率和传输效率[3]。减少排斥力的影响并抑制离子羽流的大小。毛细管和采样孔之间的距离减小会增加离子羽流密度,提高分析物的电离效率和传输效率[3]。
如Szerkus及其同事分析尿液中7-甲基鸟嘌呤和葡萄糖醛酸所证明的,上述电离源参数的优化可能会带来2到3倍的灵敏度增益[8]。在优化源条件时,使用预期的LC流动相和流速非常重要。一种优化方法是多次注入标准溶液,并在每次注射时逐步改变特定的源参数。图2展示了评估两种农药*去溶剂化温度的过程:甲an磷和甲氨基阿维菌素B1a苯甲酸盐。通过将去溶剂化温度从400℃提高到550℃,使甲an磷的响应增加20%。相反,如果去溶剂化温度超过500°C,由于该化合物的热不稳定性,甲氨基阿维菌素苯甲酸盐B1a经历*信号损失。或者,可以通过将恒定流量的分析物引入LC洗脱液并监测分析物TIC来优化源条件。该技术允许在运行中进行调整。使用多种化合物的梯度洗脱方法应通过估算洗脱时的有机物浓度来优化。尽管这一步骤势不可挡,但只需将工作集中在关键或低强度分析物上即可简化该过程。该技术允许在运行中进行调整。使用多种化合物的梯度洗脱方法应通过估算洗脱时的有机物浓度来优化。尽管这一步骤势不可挡,但只需将工作集中在关键或低强度分析物上即可简化该过程。该技术允许在运行中进行调整。使用多种化合物的梯度洗脱方法应通过估算洗脱时的有机物浓度来优化。尽管这一步骤势不可挡,但只需将工作集中在关键或低强度分析物上即可简化该过程。
图2 在四次连续注射中,(a)甲an磷和(b)甲氨基阿维菌素B1a苯甲酸酯的去溶剂化温度的LC-MS / MS优化。柱:100mm×2.1mm,3μm全多孔C18; 流动相A:水+ 2mM乙酸铵+ 0.1%甲酸; 流动相B:甲醇+ 2mM乙酸铵+ 0.1%甲酸; 梯度%B(时间):5%(0分钟),5%(1.5分钟),70%(6分钟),70%(9分钟),100%(10分钟),100%(12分钟),平衡; 流速:0.5 mL / min; 极性:ESI +; 幕气:30 psi; 雾化器气体:45 psi; 干燥气体:55 psi; 毛细管电压:5.5 kV; 碰撞气体:10 psi。 |
样品预处理
样品预处理是LC-MS分析工作流程的重要组成部分,特别是在分析含有低浓度目标分析物的复杂样品时。去除非目标样品组分可以小化基质干扰并改善目标分析物的S / N比。与目标分析物共洗脱的基质化合物可能导致分析物信号的抑制或增强; 这些干扰称为矩阵效应。基质效应通常表现为MS灵敏度或特异性的损失,并且在ESI中普遍存在,因为在发射气相离子之前可能在液滴表面上发生电荷竞争。作为替代方案,如果感兴趣的分析物是热稳定的并且具有中等极性[1],则可以使用大气压化学电离(APCI)。在APCI中,通过电晕针的施加电压,LC洗脱液在电离之前*蒸发成气体。然后电离的流动相蒸气与分析物分子反应产生带电离子。基质效应在APCI中往往不那么广泛,因为离子是通过气相反应而不是液相反应产生的[9]。
可以使用各种样品制备策略从潜在的干扰基质组分中提取目标分析物。适当的技术取决于样品基质,样品体积,目标分析物浓度和分析物理化学性质。如果样品清洁并且已知含有高浓度的目标分析物,简单的过滤和稀释是降低潜在干扰浓度的快捷方便的方法。另一方面,已知含有低目标分析物浓度的复杂样品将需要更严格的提取程序以改善信号强度。虽然由于需要投入的成本和时间,可能不需要更严格的样品制备程序,无论选择哪种样品制备技术,重要的是要考虑基质效应可能是由于内源性或外源性物质的存在。尽管样品中已存在内源性成分(蛋白质,脂质,色素等),但在样品预处理过程中将外源化合物引入样品中。这些化合物可以从用于离心管,孔板和移液管的塑料中浸出,并且可以包括来自制造过程的副产物和残余物(例如,模塑剂,增塑剂,稳定剂和释放剂)。污染物的数量和类型因制造商而异,如图3a所示。在这个实验中,对7家制造商的聚合物固相萃取(SPE)反相96孔板提取的污染物进行了比较。通过LC-MS分析提取物,并将得到的数据减去背景以除去溶剂和分析柱的贡献。所得色谱图的叠加显示各制造商之间存在多种化学污染物。基于由44Da分开的一系列重复离子,在制造商C中清楚地识别聚乙二醇(PEG)的光谱(图3b)。所得色谱图的叠加显示各制造商之间存在多种化学污染物。基于由44Da分开的一系列重复离子,在制造商C中清楚地识别聚乙二醇(PEG)的光谱(图3b)。所得色谱图的叠加显示各制造商之间存在多种化学污染物。基于由44Da分开的一系列重复离子,在制造商C中清楚地识别聚乙二醇(PEG)的光谱(图3b)。
图3 用聚合物固相萃取反相96孔板用乙腈提取的污染物,并通过LC-MS / MS分析:(a)来自七个制造商的背景减去TIC的叠加。(b)从位于6.5-8分钟的峰C收集的平均光谱。柱:100mm×2.1mm,2.7μm表面多孔C18; 流动相A:水+ 1mM乙酸铵+ 1%乙酸; 流动相B:甲醇; 梯度%B(时间):5%(0分钟),100%(8分钟),100%(9分钟),平衡; 流速:0.5 mL / min。[10] |
流动相组成
流动相通过影响目标分析物的保留和电离,在LC-MS灵敏度中发挥关键作用。使用高纯度溶剂和添加剂对于防止不需要的加合物形成和增加的MS背景是至关重要的。同样,只有来自水净化系统的超纯水或适用于LC-MS的瓶装水才能用于流动相制备。收集的MS级和HPLC级甲醇的LC-MS谱显示HPLC级甲醇中的杂质显着增加,特别是在小分子分析中常见的低分子量范围内(图4)。从该数据可以明显看出,较低等级溶剂的使用如何有助于降低灵敏度和卷积光谱,使得定量或光谱解释变得困难。
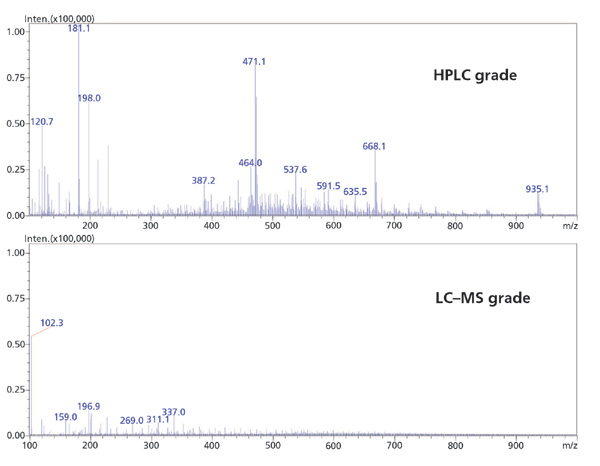 图4 HPLC级甲醇和LC-MS级甲醇的平均光谱的比较。流动相:未指明的未改性甲醇; 流速:0.5 mL / min; 系统:采用ESI +电离的LC-MS; 扫描范围:100-2000 m / z。 |
将挥发性缓冲液和酸结合到流动相中使得能够控制目标分析物的电离状态,从而可以操纵保留。分析物保留为LC-MS分析人员提供了几个优势。,增加的分析物保留意味着在梯度LC期间从柱中洗脱分析物需要更高的有机溶剂浓度。已经表明,具有较高有机浓度的液滴在MS源中更有效地去溶剂化,导致MS灵敏度提高[11]。其次,更高的色谱选择性使得可以避免可能对分析物响应有害的共洗涤基质效应。通过同时输注分析物后柱,同时通过分析柱[12]进行LC注入提取的空白基质样品,可以色谱监测保留区域和基质抑制。基质抑制区域的特征在于分析物信号的减少。通过这种方式,可以调整分析物保留,以避免色谱图中显着抑制的区域。
流动相缓冲液和酸也会影响电离效率。这种说法对于ESI尤其如此,因为由于电离竞争,它易于降低检测器响应。为了降低缓冲液诱导抑制的可能性,通常应将浓度保持在低限度。或者,含有甲酸的流动相可以使不需要的金属加合物小化。由酸提供的质子过量驱使大部分离子形成质子化分子[M + H] +,导致响应的总体改善,因为它不再分布在多个带电物质上[12]。
通过在酸离子改性剂的正离子模式下提供质子或通过在负离子模式下接受基本改性剂的质子,观察到电离效率的提高。后者被证明是两种中性雌激素,雌酮和雌三醇的负电离,当它们在含有0.2%氢氧化铵的稀释剂中制备时,与含有0.2%乙酸的雌激素相比,它们的响应增加三倍[14]。含氨的缓冲盐(例如,甲酸铵或乙酸铵)可以提高极性中性化合物的电离效率,所述极性中性化合物不能通过形成铵加合物而自身电离。铵盐可用于通过提供恒定的铵供应来防止形成不需要的加合物。例如,两种强心苷的LC-MS分析,地gao辛和洋地黄毒苷几乎*用甲酸铵改性的流动相进行。没有甲酸铵,这些化合物倾向于形成钠加合物,当通过串联MS分析时难以破碎[14]。
LC柱特性
对提高LC-MS灵敏度的渴望趋向于使用更小的颗粒(亚2μm)和减小的柱直径(≤2.1mm)实施LC柱。与全多孔颗粒(FPP)相比,表面多孔颗粒(SPP)的引入允许提率,同时降低系统压力。从理论上讲,柱可以提高灵敏度; 但是,必须考虑LC-MS系统的柱外体积,电离效率和数据采样率,以充分实现其优势。
色谱柱提供窄色谱峰的能力表征为其效率(N),并由其板高(H)定义。峰的效率是其宽度和保留时间的函数。有几个过程有助于色谱柱内外的峰展宽。进样器,连接管和检测器都是柱外峰展宽的来源。
在色谱柱内部,涡流扩散(A),纵向传质(B)以及流动相和固定相传质(C)都有助于峰的色散。总的来说,这些术语构成了van Deemter等式:

其中h是降低板高度,v是流动相线速度[2]。van Deemter方程作为比较柱性能的基础。
提高柱效的一种方法是减小粒径。减小总峰宽将导致峰高的整体增加。假设探测器噪声保持不变,较高的峰值会导致S / N的改善和灵敏度的提高。此外,峰可能更加分辨,降低了基质干扰影响电离效率的可能性。
较小的颗粒柱还允许使用更快的*线速度,并且通过扩展,更快的流速 - 不会经历效率的显着损失。不幸的是,由于控制ESI的机制,更快的流速通常不利于灵敏度,因为必须除去所有洗脱液才能成功形成气相离子。虽然一些制造商声称仪器兼容洗脱液流速高达1 mL / min,但据报道标准流量LC-ESI-MS系统的*性能发生在10-300μL/ min[16]的范围内。为了适应小颗粒及其相关的高线速度,2.1 mm内径色谱柱已成为标准流量LC-ESI-MS系统的首xuan尺寸,*流速为200-300μL/ min。
改变颗粒形态是提高柱效的另一种方法。表面多孔颗粒与全多孔颗粒的不同之处在于它们具有围绕实心核的薄多孔壳。它们能够显着提率,因为纵向扩散(B)和涡流扩散(A)的减少是由于它们的粒径分布窄,渗透性降低和外表面粗糙[17]。图5使用van Deemter图比较了全多孔3-μmC18色谱柱与相同尺寸的表面多孔2.7-μmC18色谱柱的动力学性能。与全多孔颗粒相比,表面多孔柱的效率提高了60%。
 图5 Van Deemter图比较3-μm全多孔C18柱和2.7-μm表面多孔C18柱之间的效率。流动相A:45%水; 流动相B:55%乙腈; 检测:光电二极管阵列,254 nm; 注射量:1μL; 样品:在25:75乙腈 - 水中制备的0.03mg / mL联苯。 |
利用内径较窄的色谱柱可zui大限度地减少分析物稀释,这在色谱分离过程中会发生。由于柱上稀释,分析物灵敏度与浓度依赖性检测器[18]的柱内径的平方成反比。因此,假设在两种情况下都可以注入相同体积的样品,理论上将2.1 mm内径色谱柱切换到0.3 mm内径色谱柱可以将灵敏度提高50倍[19]。同样,较小的内径柱保持相同的线速度和降低的流速,这在电离效率方面是有益的。在这些流量下,对于需要高灵敏度且样品量有限的应用,使用非常慢的流速(每分钟纳升)已经变得流行。
将窄孔柱与质谱联用时,有几个含义。柱内径的减小与效率的提高一起导致峰值体积的显着降低。在不使仪器中的柱外体积小化的情况下,柱性能将受到损害,使得难以实现灵敏度的任何显着增加。大多数柱外体积贡献可归因于LC系统,MS贡献可忽略不计。然而,发现用于将LC柱与MS系统连接的管道是关键的,因为该管道位于柱后,其中不会发生补偿谱带展宽的聚焦效应[20]。根据经验,柱外体积不应超过色谱图中窄峰的峰值体积的三分之一[21]。例如,1.8μm,100 mm×2.1 mm色谱柱的峰容积约为8μL[16]。因此,zui大柱外体积应小于3μL,以抵消系统相关的效率损失。
较小的峰值体积也意味着需要快速采集速率来收集定量数据所需峰值的小15-20个数据点。与时间相关的谱带展宽效应可能是由于停留时间不足和数据平滑过度造成的。在图6中,使用三种扫描速率(300ms,50ms和5ms)分析吗啡和氢吗啡酮。人工展宽对于300毫秒的数据是明显的,而5毫秒的数据显示过度采样的过度噪声。不正确的停留时间设置会对数据质量和信噪比产生深远的影响。此外,在分析大量化合物时,通过在选择离子监测(SIM)或多反应监测(MRM)模式下收集数据可以实现更长的循环时间,以减少与时间相关的谱带展宽效应的发生。
 图6 采集的数据与(a)300 ms,(b)50 ms和(c)5 ms的停留时间的比较,以及它们对时间相关频带展宽的贡献。系统:LC-MS / MS; 极性:ESI +。峰值:1 =吗啡,2 =氢吗啡酮。 |
结论
开发灵敏且稳健的LC-MS方法是一项艰巨的任务。为了解其目标分析物的物理化学性质,以及MS电离和传输效率的机制和局限性,分析人员可以开始做出明智的决策,以优化总体响应。提高灵敏度的简单,zui有效的方法是优化电离源条件,以确保zui大限度地生产和将气相离子转移到MS系统中。可以通过使用、窄孔LC柱,较慢的LC流速,仔细选择样品预处理程序改善响应以及减少可能导致基质效应和基线噪音的干扰从而降低检测限。
参考
[1] R.K. Boyd, C. Basic, and R.A. Bethem, Trace Quantitative Analysis by Mass Spectrometry, 1st Edition (John Wiley & Sons Ltd, West Sussex, England, 2008), pp. 242, 249.
[2] L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid Chromatography, 3rd Edition (John Wiley & Sons, Hoboken, New Jersey, 2010), pp. 39–45, 157.
[3] J.S. Page, R.T. Kelly, K. Tang, and R.D. Smith, J. Am. Soc. Mass Spectrom. 18, 1582–1590 (2007).
[4] G.I. Taylor, Proc. R. Soc. Lond. A. 280, 383 (1964).
[5] M. Wilm, Mol Cell Proteomics 10, 1–8 (2011).
[6] A. Kiontke, A. Oliveira-Birkmeier, A. Opitz, and C. Birkemeyer, PLoS One 11, 1–16 (2016).
[7]T. Taylor, LCGC Blog (7 November, 2017).
[8]O. Szerkus, A.Y. Mpanga, M.J. Markuszewski, R. Kaliszan, and D. Siluk, Spectroscopy 14, 8–16 (2016).
[9] R. Dams, M.A. Huestis, W.E. Lambert, and C.M. Murphy, J. Am. Soc. Mass Spectrom. 14, 190–1294 (2003).
[10] Y. Hua and D. Jenke, J. of Chromatogr. Sci. 50, 213-227 (2012).
[11] S.R. Needham, P.R. Brown, K. Duff, and D. Bell, J. Chromatogr. A 869, 159-170 (2000).
[12]R. Bonfiglio, R.C. King, T.V. Olah, and K. Merkle, Rapid Commun. Mass Spectrom. 13, 1175–1185 (1999).
[13]F. Klink, MS Solutions #3.
[14]S. Lupo and T. Kahler, LCGC North America 35, 424–433 (2017).
[15]J. Boertz, X. Lu, H. Brandes, S. Squillario, D. Bell, and W. Way, Poster Session presented at the Annual Meeting of the German Society for Mass Spectrometry (DGMS), Wuppertal, Germany (2015).
[16]S. Buckenmaier, C.A. Miller, T. van de Goor, and M.M. Dittman, J. Chromatogr. A 1377, 64–74 (2015).
[17]G. Guiochon and F. Gritti, J. Chromatogr. A 1218, 1915–1938 (2011).
[18]J.P.C. Vissers, H.A. Classens, and C.A. Cramers, J. Chromatogr. A 779,1–28 (1997).
[19]J. Abian, A.J. Oosterkamp and E. Gelpi, J. Mass. Spectrom. 34, 244–254 (1999).
[20]D. Spaggiari, S. Fekete, P.J. Eugster, J. Veuthey, L. Geiser, S. Rudaz, and D. Guillarme, J. Chromatogr. A 1310, 45–55 (2013).
[21]A.J. Alexander, T.J. Waeghe, K.W. Himes, F.P. Tomasella, and T.F. Hooker, J. Chromatogr. A 1218, 5456–5469 (2011).
 扫码加微信
扫码加微信

